蛋白同源性比对结果,如何实现从海量数据到精准洞察?
每天面对成百上千条BLAST或HMMER输出的比对结果,手动筛选、去重、验证,不仅耗时巨大,稍有不慎就可能遗漏那些隐藏在“灰色地带”的关键远缘同源关系。IDC的调研显示,生物学研究人员平均花费约40%的时间在数据整理而非核心分析上。这不仅拖慢了研究进度,更让创新发现被埋没在数据洪流中。本文将为你拆解一套从局部信号挖掘、多维度校验到动态报告生成的自动化整理新范式,让你重掌科研主动权。
- 挖掘“深海信号”:如何在序列相似度极低的“黄昏区”精准捕获功能指纹?
- 建立“仲裁法庭”:如何让多智能体协同工作,交叉验证,剔除假阳性?
- 交付“活态报告”:如何将整理结果变为可追溯、可交互、可自我进化的知识?
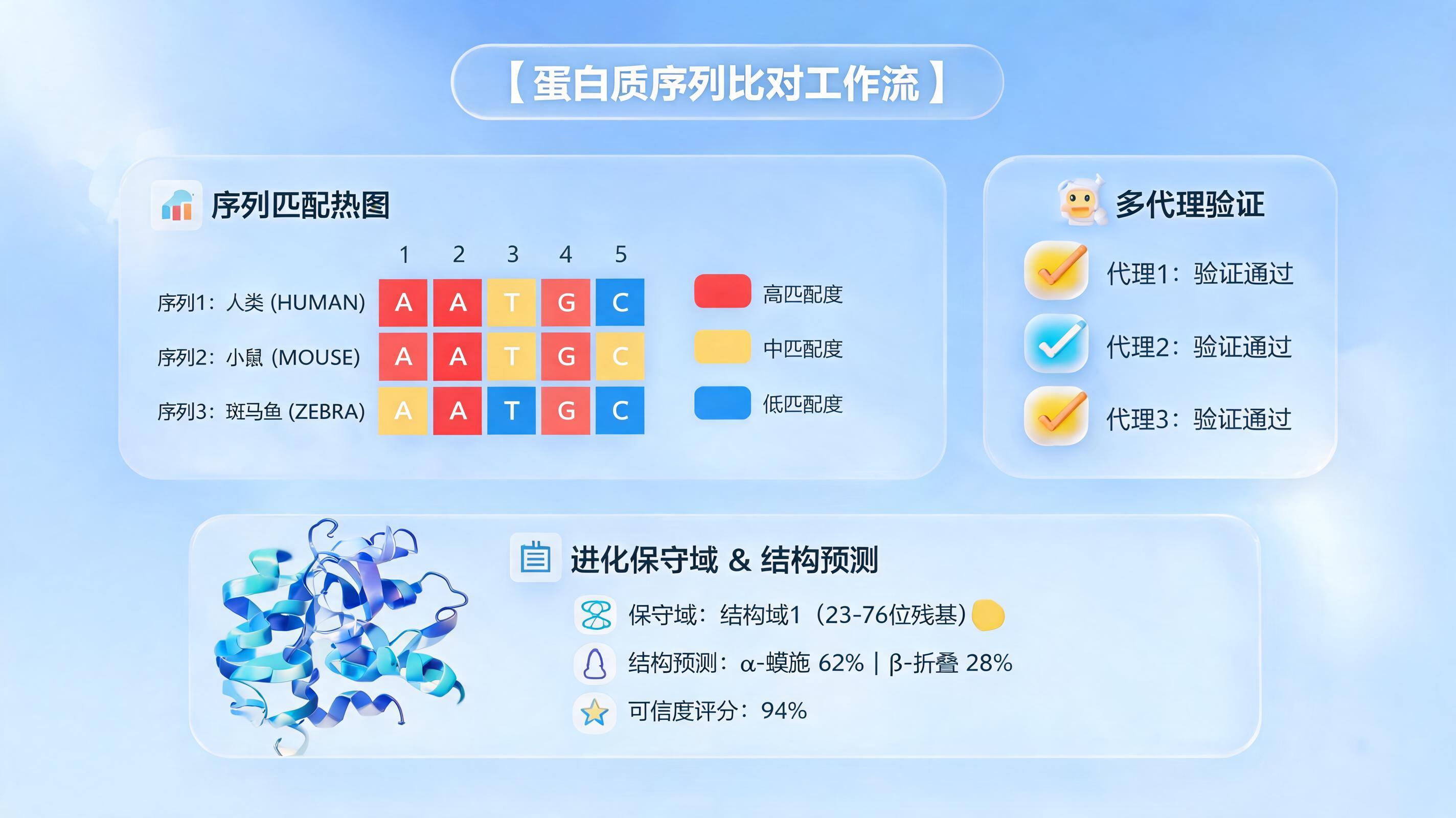
重新定义同源检索:从全局相似到局部语义匹配
传统同源比对工具在面对序列一致性低于25%的“twilight zone”时,常因过度关注全局序列相似性而失效。真正的功能相关性,往往隐藏在局部保守的结构域或关键残基中,自动整理的首要任务便是精准捕获这些“功能指纹”。
1.1 突破“黄昏区”的技术范式转移
2026年上半年的技术突破,特别是类似ProtoCol模型的提出,为我们指明了新方向。它不再将蛋白序列压缩为一个单一向量,而是借鉴延迟交互思想,为每个残基保留独立的“语义令牌”。这使得自动化系统能以一种前所未有的细粒度进行比对。
- 残基因果链构建:系统为每个残基生成嵌入,建立索引。当查询一个未知蛋白时,其每个残基都会在候选蛋白中寻找最佳局部匹配片段,执行MaxSim操作。
- 功能基序自动捕获:这种方法天然捕捉进化中保守的基序和结构域,即使它们被大片不相关序列包围,也能被精准识别。
- 假阴性结果打捞:在SCOPe superfamily等基准测试中,这种方法显著优于传统工具。对于自动化整理系统而言,集成此类算法意味着能够从以往被丢弃的垃圾数据中,重新挖掘出极具价值的远缘同源关系。
1.2 实在Agent的流程化编排应用
你可以轻松地在实在Agent中调用这类最新算法,无需编写复杂代码。通过拖拽式界面,将序列输入、远缘检索智能体、结果解析步骤编排成一个自动化流程,实在Agent会自动调度后台的GPU资源完成计算,并将原本无法识别的远缘同源蛋白挖掘出来,直接推送到你的报告草稿中。
构建协同校验机制:多智能体仲裁下的置信度评估
当多路并行的同源检索工具返回大量、甚至相互矛盾的结果时,单纯依靠规则已无法应对这种复杂性。一套模拟科研团队分工协作的智能数据治理框架,是确保最终结论可靠性的关键。
2.1 多智能体协同判断逻辑
一个理想系统的核心,是一个“仲裁”模块。面对一个仅有25%序列一致性,但由ProtoCol给出的高置信度激酶结构域匹配,系统不会简单地采信或拒绝,而是启动一个多维度校验流程。
- 分工并行验证:一个智能体负责核验比对的统计显著性(E-value),另一个解析局部匹配的生物学上下文(如是否与ATP结合位点重叠),第三个则交叉查验UniProt、PDB等公共数据库中的功能注释。
- 加权决策融合:通过加权投票或逻辑推理,系统最终给出一个综合性判断,例如,“该蛋白可能为远缘激酶家族成员”,并附带每个纬度的置信度评分和证据来源。
- 消除算法偏见:这种多智能体协同机制,有效避免了单一算法可能产生的系统性假阳性,也绕开了纯规则引擎的覆盖盲区。
2.2 实在Agent的任务树与可视化决策
借助实在Agent的“任务树”与“多智能体调度”模式,你可以将上述复杂的校验逻辑固化为标准流程。不同的校验智能体能够在实在Agent的调度下并发工作,并在“仲裁”节点汇聚。在可视化面板中,你可以清晰看到每一步推导过程,并对智能体的判断逻辑进行人工干预和微调,确保最终结论的可靠性。
交付“活”的报告:语义归一化与动态知识更新
整理的最终产出不应是死板的文本,而是一个可溯源、可交互、并能随数据更新而自我演进的“活态报告”。这要求系统具备强大的后处理、可视化以及上下文整合能力。
3.1 动态语义归一化与数据整合
面对不同数据库表述不一的情况,如“PF00069”与“SM00220”,一个高效的自动整理系统应能自动进行语义推断和归一化处理。
- 语义层级自动对齐:系统根据分析目标,自动推断不同表述的语义层级,并将它们统一归类到“蛋白激酶催化结构域”这一核心概念下。
- 多维数据维度注入:更重要的是,系统能将升级后的AlphaFold复合物结构数据纳入整理。当发现查询蛋白与某个已知同源二聚体同源后,系统会自动推断其可能的存在形式,并分析互作界面,实现从静态注释到动态功能推断的跃升。
- 标准化输出与溯源:在最终报告中,数据以标准化格式呈现,但每条数据的原始来源、算法权重和时间衰减因子都会被完整保留,形成完整的证据链。
3.2 从静态报表到洞察生成器
实在Agent的“数据处理流水线”在此能发挥巨大作用。它能将BLAST的文本输出、HMMER的DOMAIN表格等原始结果,自动转化为图形化摘要。
- 可视化后处理:通过集成Python脚本或内置的图表组件,实在Agent能直接生成“残基-残基”匹配热图,直观展示保守区域;或调用结构预测模型,生成3D交互式视图,高亮显示同源区域和结合界面。
- 可演进的分析闭环:当你对报告中的“某个远缘同源关系”提出疑问时,实在Agent能将此追问转化为新任务,自动检索突变数据库(如ClinVar)或调用亲和力预测模型(如PBCNet2.0),重新进入分析循环并更新报告,让知识始终保持最新。
这些从前需要不同团队、多个软件协作才能完成的复杂验证,现在通过一个实在Agent即可构建出完整的自动化闭环。
总结:迈向智能驱动的研究新时代
蛋白同源性比对的自动整理,已从简单的脚本过滤,进化为一个由深层语义理解、多智能体协作和动态知识图谱构成的生态系统。这套新范式不仅将研究者从繁重的数据劳作中解放出来,更重要的是,它通过挖掘远缘同源和复合物互作等深层信息,为我们打开了功能基因组学与药物靶点发现的新可能。
实在Agent正是这一范式的理想实践平台。它通过零代码的编排能力,让你能够轻松构建从序列输入到活态报告生成的自动化流程,将多模型调度、非结构化数据处理与智能决策完美融合,成为你科研探索中得力的数字员工。
常见问题解答(FAQs)
Q:BLAST结果中E-value如何确定阈值?
A:无固定标准,需结合研究目的。保守注释可用1e-10,远缘同源挖掘可放宽至1e-3甚至更高。关键要结合后续的多维度验证(保守基序、结构预测)综合判断,而非单纯依赖E-value。实在Agent支持动态设置阈值并自动分级。
Q:如何自动比对几十个蛋白质序列并整理成表格?
A:通过实在Agent构建一个“批量序列比对”流水线。输入序列列表后,系统自动依次调用Clustal Omega或MAFFT程序,运行结束后自动解析比对结果,并按一致性、空位率等指标生成标准化的对比表格,省去逐个手动操作。
Q:AlphaFold预测的结构可信吗,如何用在比对整理中?
A:AlphaFold数据库中的高置信度(pLDDT>90)结构非常可信,可用于精细地验证序列比对结果,识别保守的结构域关键残基。可靠的结构比对比序列比对更具进化上的说服力,能显著提升整理的准确度。

本文内容通过AI工具匹配关键字智能整合而成,仅供参考,实在智能不对内容的真实、准确或完整作任何形式的承诺。如有任何问题或意见,您可以通过联系contact@i-i.ai进行反馈,实在智能收到您的反馈后将及时答复和处理。